Practical problem solving
So far we have been learning about computing and coding.
The purpose of this chapter is to introduce some practical techniques for tackling data analysis problems.
At this point we go back to looking at the Swiss-Prot FASTA file used in
First steps towards automation. You can download this file using
the curl command below.
$ curl --location --output uniprot_sprot.fasta.gz http://bit.ly/1l6SAKb
The downloaded uniprot_sprot.fasta.gz file is gzipped. You can view the
content of this file without permanently unzipping it using the command below.
$ gunzip -c uniprot_sprot.fasta.gz
If the curl and gunzip commands above look unfamiliar you may want to
review the First steps towards automation chapter.
By briefly inspecting some random FASTA description lines it becomes clear that some species include variants. Below are some sample E. coli extracts from the FASTA file.
...IraP OS=Escherichia coli (strain SE11) GN=iraP PE=3 SV=1
...IraP OS=Escherichia coli (strain SMS-3-5 / SECEC) GN=iraP PE=3 SV=1
...IraP OS=Escherichia coli (strain UTI89 / UPEC) GN=iraP PE=3 SV=1
How many variants are there per species? Do all variants contain the same number of proteins?
To answer these questions we will make use of the Python scripting language and some practical problem solving techniques.
Stop and think
One of the most common mistakes when programming is to start coding before the problem is clearly defined.
What do we want to achieve? Pause for a second and try to answer this question to yourself. It may help trying to explain it out loud.
Now describe all the steps required to achieve this goal. Again it may help speaking out loud to yourself or imagining that you are describing the required steps to a colleague.
Did you find that easy or difficult? Thinking about what one wants to achieve and all the steps required to get there can often be quite overwhelming. When this happens it is easy to think to oneself that the detail will become clear later on and that one should stop wasting time and get on with the coding. Try to avoid doing this. Rather, try to think of a simpler goal.
For example consider the goal of working out how many different species there are in the FASTA file. This goal would require us to:
- Identify all the description lines in the FASTA file
- Extract the organism name from the line
- Ensure that there are no duplicate entries
- Count the number of entries
If you know the tools and techniques required to do all of the steps above this may not seem overwhelming. However, if you don’t it will. In either case it is worthwhile to treat each step as a goal in itself and work out the sub-steps required to achieve these. By iterating over this process you will either get down to steps sizes that do not feel overwhelming or you will identify the key aspect that you feel unsure about.
For example, the first step to identify all the description lines in the FASTA file can be decomposed into the sub-steps below.
- Create a list for storing the description lines
- Iterate over all the lines in the input file
- For each line check if it is a description line
- If it is add the line to the list
Now suppose one felt unsure about how to check if a line was a description line. This would represent a good place to start experimenting with some actual code. Can we come up with a code snippet that can check if a string is a FASTA description line?
Measuring success
Up until this point we have been testing our code snippets by running the code and verifying the results manually. This is terribly inefficient and soon breaks down once you have more than one thing to test.
It is much better to write tests that can be run automatically. Let’s examine
this concept. Create a file named fasta_utils.py and add the Python
code below to it.
1 2 3 4 5 6 7 | """Module containing utility functions for working with FASTA files."""
def test_is_description_line():
"""Test the is_description_line() function."""
print("Testing the is_description_line() function...")
test_is_description_line()
|
The first line is a module level “docstring” and it is used to explain the
intent of the module as a whole. A “module” is basically a file with a .py
extension, i.e. a Python file. Modules are used to group related functionality
together.
Lines three to five represent a function named test_is_description_line().
Python functions consist of two parts: the function definition (line 3) and the
function body (lines 4 and 5).
In Python functions are defined using the def keyword. Note that the
def keyword is followed by the name of the function. The name of the
function is followed by a parenthesized set of arguments, in this case the
function takes no arguments. The end of the function definition is marked using
a colon.
The body of the function, lines four and five, need to be indented. The standard in Python is to use four white spaces to indent code blocks.
Avertissement
Whitespace really matters in Python! If your code is not correctly
aligned you will see IndentationError messages telling you
that everything is not as it should be. You will also run into
IndentationError messages if you mix white spaces and tabs.
The first line of the function body, line four, is a docstring explaining the
intent of the function. Line five makes use of the built-in print()
function to write a string to the standard output stream. Python’s built-in
print() function is similar to the echo command we used earlier in
Keeping track of your work.
Finally, on line seven the test_is_description_line() function is called,
i.e. the logic within the function’s body is executed. In this instance this
means that the "Testing the is_description_line() function..." string is
written to the standard output stream.
Let us run this script in a terminal. To run a Python script we use the python
command followed by the name of the script.
$ python fasta_utils.py
Testing the is_description_line() function...
So far so good? At the moment our test_is_description_line() function
does not actually test anything. Let us rectify that now.
1 2 3 4 5 6 7 8 | """Module containing utility functions for working with FASTA files."""
def test_is_description_line():
"""Test the is_description_line() function."""
print("Testing the is_description_line() function...")
assert is_description_line(">This is a description line") is True
test_is_description_line()
|
There are quite a few things going on in the newly added line. First of all it
makes use of three built-in features of Python: the assert and is
keywords, as well as the True constant. Let’s work through these in reverse
order.
Python has some built-in constants, most notably True,
False and None. The True and False constants are the only
instances of the bool (boolean) type and None is often used to
represent the absence of a value.
In Python is is an operator that checks for object identity, i.e. if the
object returned by the is_description_line() function and True are
the same object. If they are the same object the comparison evaluates to
True if not it evaluates to False.
The assert keyword is used to insert debugging statements into a program.
It provides a means to ensure that the state of a program is as expected. If
the statement evaluates to False an AssertionError is raised.
So, what will happen if we run the code in its current form?
Well, we have not yet defined the is_description_line() function, so
Python will raise a NameError. Let us run the code.
$ python fasta_utils.py
Testing the is_description_line() function...
Traceback (most recent call last):
File "fasta_utils.py", line 8, in <module>
test_is_description_line()
File "fasta_utils.py", line 6, in test_is_description_line
assert is_description_line(">This is a description line") is True
NameError: global name 'is_description_line' is not defined
Great now we are getting somewhere! What? Well, we have impemented some
code to test the functionality of the is_description_line() and it
tells us that the function does not exist. This is useful information.
Let us add a placeholder is_description_line() function to the Python
module.
1 2 3 4 5 6 7 8 9 10 11 | """Module containing utility functions for working with FASTA files."""
def is_description_line(line):
"""Return True if the line is a FASTA description line."""
def test_is_description_line():
"""Test the is_description_line() function."""
print("Testing the is_description_line() function...")
assert is_description_line(">This is a description line") is True
test_is_description_line()
|
Note that the function we have added on lines three and four currently does nothing.
By default the function will return None. However, when we run the script
we should no longer get a NameError. Let’s find out what happens when we
run the code.
$ python fasta_utils.py
Testing the is_description_line() function...
Traceback (most recent call last):
File "fasta_utils.py", line 11, in <module>
test_is_description_line()
File "fasta_utils.py", line 9, in test_is_description_line
assert is_description_line(">This is a description line") is True
AssertionError
More progress! Now we see the expected AssertionError, becuase None is
not True. Let us add some code to try to get rid of this error message. To
achieve this we simply need to make the function return True.
1 2 3 4 5 6 7 8 9 10 11 12 | """Module containing utility functions for working with FASTA files."""
def is_description_line(line):
"""Return True if the line is a FASTA description line."""
return True
def test_is_description_line():
"""Test the is_description_line() function."""
print("Testing the is_description_line() function...")
assert is_description_line(">This is a description line") is True
test_is_description_line()
|
Now, we can run the code again.
$ python fasta_utils.py
Testing the is_description_line() function...
No error message, the code is now working to the specification described in the test. However, the test does not specify what the behaviour should be for a biological sequence line. Let us add another assert statement to specify this.
1 2 3 4 5 6 7 8 9 10 11 12 13 | """Module containing utility functions for working with FASTA files."""
def is_description_line(line):
"""Return True if the line is a FASTA description line."""
return True
def test_is_description_line():
"""Test the is_description_line() function."""
print("Testing the is_description_line() function...")
assert is_description_line(">This is a description line") is True
assert is_description_line("ATCG") is False
test_is_description_line()
|
Now we can run the code again.
$ python fasta_utils.py
Testing the is_description_line() function...
Traceback (most recent call last):
File "fasta_utils.py", line 13, in <module>
test_is_description_line()
File "fasta_utils.py", line 11, in test_is_description_line
assert is_description_line("ATCG") is False
AssertionError
More progress, we now have a test to ensure that the is_description_line() function
returns False when the input line is a sequence. Let us try to implement the desired
functionality to make the test pass. For this we will use the
startswith() method,
that is built into strings, to check if the string starts with a greater than (>) character.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | """Module containing utility functions for working with FASTA files."""
def is_description_line(line):
"""Return True if the line is a FASTA description line."""
if line.startswith(">"):
return True
else:
return False
def test_is_description_line():
"""Test the is_description_line() function."""
print("Testing the is_description_line() function...")
assert is_description_line(">This is a description line") is True
assert is_description_line("ATCG") is False
test_is_description_line()
|
In the code above we make use of conditional logic, i.e. if something is
True the code does something otherwise it does something else. As mentioned previously
whitespace is important in Python and four spaces are used to indent the lines after
the if and else statements to tell Python which statement(s) belong in the conditional
code blocks. In this case we only have one statement per conditional, but it is
possible to group several statements together based on their indentation.
Let us test the code again.
$ python fasta_utils.py
Testing the is_description_line() function...
Fantastic the code behaves in the way that we want it to behave!
However, the current implementation of the is_description_line() function
is a little bit verbose. Do we really need the else conditional? What
would happen if it was not there?
The beauty of tests now become more apparent. We can start experimenting with the implementation of a function and feel confident that we are not breaking it. As long as the tests do not fail that is!
Let us test out our hypothesis that the else conditional is redundant by
removing it and de-denting the return False statement.
1 2 3 4 5 6 7 | """Module containing utility functions for working with FASTA files."""
def is_description_line(line):
"""Return True if the line is a FASTA description line."""
if line.startswith(">"):
return True
return False
|
Now we can simply run the tests to see what happens.
$ python fasta_utils.py
Testing the is_description_line() function...
Amazing, we just made a change to our code and we can feel pretty sure that it is
still working as intended. This is very powerful. Incidentally, the code works
by virtue of the fact that if the return True statement is reached the function
returns True and exits, as such the second return statement is not called.
The methodology used in this section is known as Test-Driven Development, often referred to as TDD. It involves three steps:
- Write a test
- Write minimal code to make the test pass
- Refactor the code if necessary
In this instance we started off by writing a test checking that the
is_description_line() function returned True when the input was a
description line. We then added minimal code to make the test pass, i.e. we
simply made the function return True. At this point no refactoring was
needed so we added another test to check that the function returned False
when the input was a sequence line. We then added some naive code to make the
tests pass. At this point, we believed that there was scope to improve the
implementation of the function, so we refactored it to remove the redundant
else statement.
Well done! That was a lot of information. Go make yourself a cup of tea.
More string processing
Because information about DNA and proteins are often stored in plain text files many aspects of biological data processing involves manipulating text. In computing text is often referred to as strings of characters. String manipulation is is therefore a common task both for processing biological sequences and for interpreting sequence identifiers.
This section will provide a brief summary of how Python can be used for such string processing.
Start Python in its interactive mode by typing python into the terminal.
$ python
Python 2.7.10 (default, Jul 14 2015, 19:46:27)
[GCC 4.2.1 Compatible Apple LLVM 6.0 (clang-600.0.39)] on darwin
Type "help", "copyright", "credits" or "license" for more information.
>>>
The Python string object
When parsing in strings from a text file one often has to deal with lines that
have leading and/or trailing white spaces. Commonly one wants to get rid of them.
This can be achieved using the strip() method built into the string object.
>>> " text with leading/trailing spaces ".strip()
'text with leading/trailing spaces'
Another common use case is to replace a word in a line. For example,
when we strip out the leading and trailing white spaces one might want to
update the word “with” to “without” to make the resulting string reflect
its current state. This can be achieved using the replace() method.
>>> " text with leading/trailing spaces ".strip().replace("with", "without")
'text without leading/trailing spaces'
Note
In the example above we chain the strip() and replace() methods
together. In practise this means that the replace() methods acts
on the return value of the strip() method.
Earlier we saw how the startswith() method can be used to identify FASTA
description lines.
>>> ">MySeq1|description line".startswith(">")
True
The endswith() method complements the startswith() method and is often
used to examine file extensions.
>>> "/home/olsson/images/profile.png".endswith("png")
True
This example above only works if the file extension is in lower case.
>>> "/home/olsson/images/profile.PNG".endswith("png")
False
However, we can overcome this issue by adding a call to the lower() method,
which converts the string to lower case.
>>> "/home/olsson/images/profile.PNG".lower().endswith("png")
True
Another common use case is to search for a particular string within
another string. For example one might want to find out if the UniProt
identifier “Q6GZX4” is present in a FASTA description line. To achieve this one
can use the find() method, which returns the index position (zero-based)
where the search term was first identified.
>>> ">sp|Q6GZX4|001R_FRG3G".find("Q6GZX4")
4
If the search term is not identified find() returns -1.
>>> ">sp|P31946|1433B_HUMAN".find("Q6GZX4")
-1
When iterating over lines in a file one often wants to split the line based on a
delimiter. This can be achieved using the split() method. By default this
splits on white space characters and returns a list of strings.
>>> "text without leading/trailing spaces".split()
['text', 'without', 'leading/trailing', 'spaces']
A different delimiter can be used by providing it as an argument to the split()
method.
>>> ">sp|Q6GZX4|001R_FRG3G".split("|")
['>sp', 'Q6GZX4', '001R_FRG3G']
There are many variations on the string operators described above. It is useful to familiarise yourself with the Python documentation on strings.
Regular expressions
Regular expressions can be defined as a series of characters that define a search pattern.
Regular expressions can be very powerful. However, they can be difficult to build up. Often it is a process of trial and error. This means that once they have been created, and the trial and error process has been forgotten, it can be extremely difficult to understand what the regular expression does and why it is constructed the way it is.
Avertissement
Use regular expression as a last resort. A good rule of thumb is to always try to use string operations to implement the desired functionality and only switch to regular expressions when the code implemented using these operations becomes more difficult to understand than the equivalent regular expression.
To use regular expressions in Python we need to import the re module.
The re module is part of Python’s standard library. Importing modules
in Python is achieved using the import keyword.
>>> import re
Let us store a FASTA description line in a variable.
>>> fasta_desc = ">sp|Q6GZX4|001R_FRG3G"
Now, let us search for the UniProt identifer Q6GZX4 within the line.
>>> re.search(r"Q6GZX4", fasta_desc)
<_sre.SRE_Match object at 0x...>
There are two things to note here:
- We use a raw string to represent our regular expression, i.e. the string prefixed with an
r - The regular expression
search()method returns a match object (or None if no match is found)
The index of the first matched character can be accessed using the match
object’s start() method. The match object also has an end() method
that returns the index of the last character + 1.
>>> match = re.search(r"Q6GZX4", fasta_desc)
>>> if match:
... print(fasta_desc[match.start():match.end()])
...
Q6GZX4
In the above we make use of the fact that Python strings support slicing.
Slicing is a means to access a subsection of a sequence. The [start:end]
syntax is inclusive for the start index and exclusive for the end index.
>>> "012345"[2:4]
'23'
To see the merit of regular expressions we need to create one that matches more
than one thing. For example a regular expression that could match all the
patterns id0, id1, ..., id9.
Now suppose that we had a list containing FASTA description lines with these types of identifiers.
>>> fasta_desc_list = [">id0 match this",
... ">id9 and this",
... ">id100 but not this (initially)",
... "AATCG"]
...
Note that the list above also contains a sequence line that we never want to match.
Let us loop over the items in this list and print out the lines that match our identifier regular expression.
>>> for line in fasta_desc_list:
... if re.search(r">id[0-9]\s", line):
... print(line)
...
>id0 match this
>id9 and this
There are two noteworthy aspects of the regular
expression. Firstly, the [0-9] syntax means match any digit.
Secondly, the \s regular
expression meta character means match any white space character.
If one wanted to create a regular expression to match an identifier with an
arbitrary number of digits one can make use of the * meta character, which
causes the regular expression to match the preceding expression 0 or more times.
>>> for line in fasta_desc_list:
... if re.search(r">id[0-9]*\s", line):
... print(line)
...
>id0 match this
>id9 and this
>id100 but not this (initially)
It is possible to extract specific pieces of information from a line using regular expressions. This uses a concept known as “groups”, which are indicated using parenthesis. Let us try to extract the UniProt identifier from a FASTA description line.
>>> print(fasta_desc)
>sp|Q6GZX4|001R_FRG3G
>>> match = re.search(r">sp\|([A-Z,0-9]*)\|", fasta_desc)
Avertissement
Note how horrible and incomprehensible the regular expression is.
It took me a couple of attempts to get this regular expression right as I
forgot that | is a regular expression meta character that needs to be
escaped using a backslash \.
The regular expression representing the UniProt idendifier [A-Z,0-9]* means
match capital letters (A-Z) and digits (0-9) zero or more times (*).
The UniProt regular expression is enclosed in parenthesis. The parenthesis
denote that the UniProt identifier is a group that we would like access to. In
other words, the purpose of a group is to give the user access to a section of
interest within the regular expression.
>>> match.groups()
('Q6GZX4',)
>>> match.group(0) # Everything matched by the regular expression.
'>sp|Q6GZX4|'
>>> match.group(1)
'Q6GZX4'
Note
There is a difference between the groups() and the group()
methods. The former returns a tuple containing all the groups
defined in the regular expression. The latter takes an integer as
input and returns a specific group. However, confusingly group(0)
returns everything matched by the regular expression and group(1)
returns the first group making the group() method appear as if
it used a one-based indexing scheme.
Finally Let us have a look at a common pitfall when using regular expressions in Python: the difference between the methods search() and match().
>>> print(re.search(r"cat", "my cat has a hat"))
<_sre.SRE_Match object at 0x...>
>>> print(re.match(r"cat", "my cat has a hat"))
None
Basically match() only looks for a match at the beginning of the string to
be searched. For more information see the
search() vs match()
section in the Python documentation.
There is a lot more to regular expressions in particular all the meta characters. For more information have a look at the regular expressions operations section in the Python documentation.
Extracting the organism name
Armed with our new found knowledge of string processing let’s create a function for extracting the organism name from a SwissProt FASTA description line. In other words given the lines:
>sp|P01090|2SS2_BRANA Napin-2 OS=Brassica napus PE=2 SV=2
>sp|Q15942|ZYX_HUMAN Zyxin OS=Homo sapiens GN=ZYX PE=1 SV=1
>sp|Q6QGT3|A1_BPT5 A1 protein OS=Escherichia phage T5 GN=A1 PE=2 SV=1
We would like to extract the strings:
Brassica napus
Homo sapiens
Escherichia phage T5
There are three things which are worth noting:
- The organism name string is always preceeded by the key
OS(Organism Name) - The organism name string can contain more than two words
- The two letter key after the organism name string can vary, in the case
above we see both
PS(Protein Existence) andGE(Gene Name)
For more information about the UniProt FASTA description line go to UniProt’s FASTA header page.
The three FASTA description lines examined above provide an excellent basis for
creating a test for the function that we want. Add the lines below
to your fasta_utils.py file.
15 16 17 18 19 20 21 22 23 24 25 | def test_extract_organism_name():
"""Test the extract_organism_name() function."""
print("Testing the extract_organism_name() function...")
lines = [">sp|P01090|2SS2_BRANA Napin-2 OS=Brassica napus PE=2 SV=2",
">sp|Q15942|ZYX_HUMAN Zyxin OS=Homo sapiens GN=ZYX PE=1 SV=1",
">sp|Q6QGT3|A1_BPT5 A1 protein OS=Escherichia phage T5 GN=A1 PE=2 SV=1"]
organism_names = ["Brassica napus", "Homo sapiens", "Escherichia phage T5"]
for line, organism_name in zip(lines, organism_names):
assert extract_organism_name(line) == organism_name
test_is_description_line()
|
In the above we make use of pythons built-in zip() function. This function takes two
lists as inputs and returns a list with paired values from the input lists.
>>> zip(["a", "b", "c"], [1, 2, 3])
[('a', 1), ('b', 2), ('c', 3)]
Let’s make sure that the tests fail.
$ python fasta_utils.py
Testing the is_description_line() function...
What, no error message, what is going on? Ah, we added the test, but forgot to add a line to call it. Let’s rectify that.
25 26 | test_is_description_line()
test_extract_organism_name()
|
Let’s try again.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Traceback (most recent call last):
File "fasta_utils.py", line 26, in <module>
test_extract_organism_name()
File "fasta_utils.py", line 23, in test_extract_organism_name
assert extract_organism_name(line) == organism_name
NameError: global name 'extract_organism_name' is not defined
Success! We now have a failing test informing us that we need to create the
extract_organism_name() function. Let’s do that.
15 16 | def extract_organism_name(line):
"""Return the organism name from a FASTA description line."""
|
Let’s find out where this minimal implementation gets us.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Traceback (most recent call last):
File "fasta_utils.py", line 29, in <module>
test_extract_organism_name()
File "fasta_utils.py", line 26, in test_extract_organism_name
assert extract_organism_name(line) == organism_name
AssertionError
The test fails as expected. However, since we are looping over many input
lines it would be good to get an idea of which test failed. We can achieve this
by making use of the fact that we can provide a custom message to be passed to
the AssertionError. Let us pass it the input line. Note the addition of the
trailing , line in line 26.
25 26 | for line, organism_name in zip(lines, organism_names):
assert extract_organism_name(line) == organism_name, line
|
Let’s see what we get now.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Traceback (most recent call last):
File "fasta_utils.py", line 29, in <module>
test_extract_organism_name()
File "fasta_utils.py", line 26, in test_extract_organism_name
assert extract_organism_name(line) == organism_name, line
AssertionError: >sp|P01090|2SS2_BRANA Napin-2 OS=Brassica napus PE=2 SV=2
Much better! Let us try to implement a basic regular expression to make this
pass. First of all we need to make sure we import the re
module.
1 2 3 | """Module containing utility functions for working with FASTA files."""
import re
|
Then we can implement a regular expression to try to extract the organism name.
17 18 19 20 | def extract_organism_name(line):
"""Return the organism name from a FASTA description line."""
match = re.search(r"OS=(.*) PE=", line)
return match.group(1)
|
Remember the .* means match any character zero or more times and the
surrounding parenthesis creates a group.
Let us see what happens now.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Traceback (most recent call last):
File "fasta_utils.py", line 34, in <module>
test_extract_organism_name()
File "fasta_utils.py", line 31, in test_extract_organism_name
assert extract_organism_name(line) == organism_name, line
AssertionError: >sp|Q15942|ZYX_HUMAN Zyxin OS=Homo sapiens GN=ZYX PE=1 SV=1
Progress! We are now seeing a different error message. The issue is that the key after
the regular expression is GN rather than PE. Let us try to rectify that.
17 18 19 20 | def extract_organism_name(line):
"""Return the organsim name from a FASTA description line."""
match = re.search(r"OS=(.*) [A-Z]{2}=", line)
return match.group(1)
|
The regular expression now states that instead of PE it wants any capital
letter [A-Z] repeated twice {2}. Let’s find out if this fixes the issue.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Traceback (most recent call last):
File "fasta_utils.py", line 33, in <module>
test_extract_organism_name()
File "fasta_utils.py", line 30, in test_extract_organism_name
assert extract_organism_name(line) == organism_name, line
AssertionError: >sp|P01090|2SS2_BRANA Napin-2 OS=Brassica napus PE=2 SV=2
What, back at square one again? As mentioned previously, regular expressions can be painful and should only be used as a last resort. This also exemplifies why it is important to have tests. Sometimes you think you make an innocuous change, but instead things just fall apart.
At this stage the error message is not very useful, let us change it to print out the value returned by the function instead.
29 30 | for line, organism_name in zip(lines, organism_names):
assert extract_organism_name(line) == organism_name, extract_organism_name(line)
|
Now, let’s see what is going on.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Traceback (most recent call last):
File "fasta_utils.py", line 33, in <module>
test_extract_organism_name()
File "fasta_utils.py", line 30, in test_extract_organism_name
assert extract_organism_name(line) == organism_name, extract_organism_name(line)
AssertionError: Brassica napus PE=2
Our regular expression is basically matching too much. The reason for this is that the
* meta character acts in a “greedy” fashion matching as much as possible,
see Fig. 20. In this case
the PE=2 is included in the match group as [A-Z]{2} is matched by the SV= key
at the end of the line. The fix is to make the * meta character act in a “non-greedy”
fashion. This is achieved by adding a ? suffix to it.
17 18 19 20 | def extract_organism_name(line):
"""Return the organism name from a FASTA description line."""
match = re.search(r"OS=(.*?) [A-Z]{2}=", line)
return match.group(1)
|
Let’s find out what happens now.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
All the tests pass! Well done, time for another cup of tea.
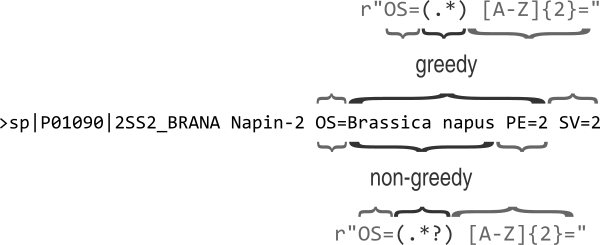
Fig. 20 Figure illustrating the difference between a greedy and a non-greedy regular
expression. The * meta-character causes the previous regular expression,
in this case the any character (.), to be matched zero or more times.
This is done in a greedy fashion, i.e. trying to match as much text as
possible. To make that part of the regular expression match as little text
as possible, i.e. to work in a non-greedy fashion, we use the meta-character
*?.
Only running tests when the module is called directly
In Python, modules provide a means to group related functionality together. For
example we have already looked at and made use of the re module, which
groups functionality for working with regular expressions.
In Python any file with a .py extension is a module. This means that the
file that we have been creating, fasta_utils.py, is a module.
To make use of the functionality within a module one needs to import it.
Let’s try this out in an interactive session
$ python
Now we can import the module.
>>> import fasta_utils
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Note that the tests run just like when we call the fasta_utils.py
script directly. This is an undesired side effect of the current
implementation. It would be better if the tests were not run when the module
is imported.
To improve the behaviour of the fasta_utils module we will make use of a
special Python attribute called __name__, which provides a string representation
of scope. When commands are run from a script or the interactive prompt the
name attribute is set to __main__. When a module is imported the __name__
attribute is set to the name of the module.
>>> print(__name__)
__main__
>>> print(fasta_utils.__name__)
fasta_utils
Using this information we can update the fasta_utils.py file with
the changes highlighted below.
32 33 34 | if __name__ == "__main__":
test_is_description_line()
test_extract_organism_name()
|
Let us make sure that the tests still run if we run the script directly.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Now we can reload the module in the interactive prompt we were working in earlier to make sure that the tests no longer get executed.
>>> reload(fasta_utils)
<module 'fasta_utils' from 'fasta_utils.py'>
The test suite is no longer being called upon loading the module and no lines logging the progress of the testing suite is being printed to the terminal.
Note that simply calling the import fasta_utils command again will not
actually detect the changes that we made to the fasta_utils.py file,
which is why we make use of Python’s built-in reload() function. Alternatively,
one could have exited the Python shell, using Ctrl-D or the exit() function,
and then started a new interactive Python session and imported the fasta_utils
module again.
Counting the number of unique organisms
We can now use the fasta_utils module to start answering some of the
biological questions that we posed at the beginning of this chapter. For now
let us do this using an interactive Python shell.
$ python
Python 2.7.10 (default, Jul 14 2015, 19:46:27)
[GCC 4.2.1 Compatible Apple LLVM 6.0 (clang-600.0.39)] on darwin
Type "help", "copyright", "credits" or "license" for more information.
>>>
Now we will start by importing the modules that we want to use. In this case
fasta_utils for processing the data and gzip for opening and
reading in the data from the uniprot_sprot.2015-11-26.fasta.gz file.
>>> import fasta_utils
>>> import gzip
Now we create a list for storing all the FASTA description lines.
>>> fasta_desc_lines = []
We can then open the file using the gzip.open() function. Note that this
returns a file handle.
>>> file_handle = gzip.open("../data/uniprot_sprot.2015-11-26.fasta.gz")
Using a for loop we can iterate over all the lines in the input file.
>>> for line in file_handle:
... if fasta_utils.is_description_line(line):
... fasta_desc_lines.append(line)
...
When we are finished with the input file we must remember to close it.
>>> file_handle.close()
Let’s check that the number of FASTA description lines using the built-in
len() function. This function returns the number of items
in the list, i.e. the length of the list.
>>> len(fasta_desc_lines)
549832
Okay now it is time to find the number of unique organisms. For this we will make
use of a data structure called set. In Python sets are used to compare
collections of unique elements. This means that sets are ideally suited for
operations that you may associate with Venn diagrams.
However, in this instance we simply use the set data structure to ensure
that we only get one unique representative of each organism. In other words
even if one calls the set.add() function several times with the same item
the item will only occur once in the set.
>>> organisms = set()
>>> for line in fasta_desc_lines:
... s = fasta_utils.extract_organism_name(line)
... organisms.add(s)
...
>>> len(organisms)
13251
Great, now we know that there are 13,251 unique organisms represented in the FASTA file.
Finding the number of variants per species and the number of proteins per variant
Suppose we had a FASTA file containing only four entries with the description lines below.
>sp|P12334|AZUR1_METJ Azurin iso-1 OS=Methylomonas sp. (strain J) PE=1 SV=2
>sp|P12335|AZUR2_METJ Azurin iso-2 OS=Methylomonas sp. (strain J) PE=1 SV=1
>sp|P23827|ECOT_ECOLI Ecotin OS=Escherichia coli (strain K12) GN=eco PE=1 SV=1
>sp|B6I1A7|ECOT_ECOSE Ecotin OS=Escherichia coli (strain SE11) GN=eco PE=3 SV=1
The analysis in the previous section would have identified these as three separate entities.
Methylomonas sp. (strain J)
Escherichia coli (strain K12)
Escherichia coli (strain SE11)
Now, suppose that we wanted to find out how many variants there were of each
species. In the example above there would be be one variant of Methylomonas
sp. and two variants of Escherichia coli. Furthermore, suppose that we
also wanted to find out how many proteins were associated with each variant.
We could achieve this by creating a nested data structure using Python’s built
in dictionary type. At the top level we should have a dictionary
whose keys were the species, e.g. Escherichia coli. The values in the top
level dictionary should themselves be dictionaries. The keys of the nested dictionaries
should be the full organism name, e.g. Escherichia coli (strain K12). The values
in the nested dictionary should be an integer representing the number of proteins found
for that organism. Below is a YAML representation of the data structure that should be
created from the four entries above.
---
Methylomonas sp.:
Methylomonas sp. (strain J): 2
Escherichia coli:
Escherichia coli (strain K12): 1
Escherichia coli (strain SE11): 1
So what type of functionality would we need to achieve this? First of all we need a
function that given an organism name returns the associated species. In other words
something that converts Escherichia coli (strain K12) to Escherichia coli.
Secondly, we need a function that given a list of organism names returns the data
structure described above.
Let us start by creating a test for converting the organism name into a species.
Add the test below to fasta_utils.py.
32 33 34 35 36 37 38 39 40 | def test_organism_name2species():
print("Testing the organism_name2species() function...")
assert organism_name2species("Methylomonas sp. (strain J)") == "Methylomonas sp."
assert organism_name2species("Homo sapiens") == "Homo sapiens"
if __name__ == "__main__":
test_is_description_line()
test_extract_organism_name()
test_organism_name2species()
|
Let’s find out what error this gives us.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Testing the organism_name2species() function...
Traceback (most recent call last):
File "fasta_utils.py", line 40, in <module>
test_organism_name2species()
File "fasta_utils.py", line 34, in test_organism_name2species
assert organism_name2species("Methylomonas sp. (strain J)") == "Methylomonas sp."
NameError: global name 'organism_name2species' is not defined
The error message is telling us that we need to define the
organism_name2species() function. Add the lines below to define it.
32 33 | def organism_name2species(organism_name):
"""Return the species from the FASTA organism name."""
|
Now we get a new error message when we run the tests.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Testing the organism_name2species() function...
Traceback (most recent call last):
File "fasta_utils.py", line 43, in <module>
test_organism_name2species()
File "fasta_utils.py", line 37, in test_organism_name2species
assert organism_name2species("Methylomonas sp. (strain J)") == "Methylomonas sp."
AssertionError
Great, let us add some logic to the function.
32 33 34 35 | def organism_name2species(organism_name):
"""Return the species from the FASTA organism name."""
words = organism_name.split()
return words[0] + " " + words[1]
|
Above, we split the organism name based on whitespace separators and return the first two words joined by a space character.
Note
In Python, and many other scripting languages, strings can be concatenated using the + operator.
>>> "hello" + " " + "world"
'hello world'
Time to test the code again.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Testing the organism_name2species() function...
Great, the function is working! Let us define a new test for the function that will generate the nested data structure we described earlier.
42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 | def test_summarise_species_protein_data():
print("Testing summarise_species_protein_data() function...")
fasta_desc_lines = [
">sp|P12334|AZUR1_METJ Azurin iso-1 OS=Methylomonas sp. (strain J) PE=1 SV=2",
">sp|P12335|AZUR2_METJ Azurin iso-2 OS=Methylomonas sp. (strain J) PE=1 SV=1",
">sp|P23827|ECOT_ECOLI Ecotin OS=Escherichia coli (strain K12) GN=eco PE=1 SV=1",
">sp|B6I1A7|ECOT_ECOSE Ecotin OS=Escherichia coli (strain SE11) GN=eco PE=3 SV=1"
]
summary = summarise_species_protein_data(fasta_desc_lines)
# The top level dictionary will contain two entries.
assert len(summary) == 2
assert "Methylomonas sp." in summary
assert "Escherichia coli" in summary
# The value of the Methylomonas sp. entry is a dictionary with one
# entry in it.
assert len(summary["Methylomonas sp."]) == 1
assert summary["Methylomonas sp."]["Methylomonas sp. (strain J)"] == 2
# The value of the Escherichia coli entry is a dictionary with two
# entries in it.
assert len(summary["Escherichia coli"]) == 2
assert summary["Escherichia coli"]["Escherichia coli (strain K12)"] == 1
assert summary["Escherichia coli"]["Escherichia coli (strain SE11)"] == 1
if __name__ == "__main__":
test_is_description_line()
test_extract_organism_name()
test_organism_name2species()
test_summarise_species_protein_data()
|
This should all be getting familiar now. Time to run the tests.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Testing the organism_name2species() function...
Testing summarise_species_protein_data() function...
Traceback (most recent call last):
File "fasta_utils.py", line 70, in <module>
test_summarise_species_protein_data()
File "fasta_utils.py", line 50, in test_summarise_species_protein_data
summary = summarise_species_protein_data(fasta_desc_lines)
NameError: global name 'summarise_species_protein_data' is not defined
Again we start by defining the function.
42 43 | def summarise_species_protein_data(fasta_desc_lines):
"""Return data structure summarising the organism and protein data."""
|
And then we run the tests again.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Testing the organism_name2species() function...
Testing summarise_species_protein_data() function...
Traceback (most recent call last):
File "fasta_utils.py", line 74, in <module>
test_summarise_species_protein_data()
File "fasta_utils.py", line 57, in test_summarise_species_protein_data
assert len(summary) == 2
TypeError: object of type 'NoneType' has no len()
Time to add an implementation.
42 43 44 45 46 47 48 49 50 51 | def summarise_species_protein_data(fasta_desc_lines):
"""Return data structure summarising the organism and protein data."""
summary = dict()
for line in fasta_desc_lines:
variant_name = extract_organism_name(line)
species_name = organism_name2species(variant_name)
variant_dict = summary.get(species_name, dict())
variant_dict[variant_name] = variant_dict.get(variant_name, 0) + 1
summary[species_name] = variant_dict
return summary
|
In the above we make use of the dictionary’s built-in get() method, which
returns the value associated with the key provided as the first argument. If the
key does not yet exist in the dictionary it returns the second argument, the
default value.
On line 48 we try to access a variant_dict dictionary from within the summary
dictionary, we are in other words working with a nested data structure. If there
is no entry associated with the species_name the get() method will return
an empty dictionary (dict()).
On line 49 we keep count of the number of times a variant of a species, as defined
by the full organism name, has been observed. In this instance we
use the get() method to get the number of times the variant has been observed
previously and then we add 1 to it. If the variant has never been observed previously
it will not be in the dictionary and the variant_dict.get(variant_name, 0) method
call will return 0 (the default value specified).
Line 50 updates the top level dictionary by adding the
variant_dict dictionary to the summary dictionary.
Let’s see if it the implementation works as expected.
$ python fasta_utils.py
Testing the is_description_line() function...
Testing the extract_organism_name() function...
Testing the organism_name2species() function...
Testing summarise_species_protein_data() function...
Hurray!
Finally, let us write a separate script to convert an input FASTA file into a
YAML summary file. Create the file fasta2yaml_summary.py and add the
code below to it.
#!/usr/bin/env python
import sys
import yaml
import fasta_utils
fasta_desc_lines = list()
with sys.stdin as fh:
for line in fh:
if fasta_utils.is_description_line(line):
fasta_desc_lines.append(line)
summary = fasta_utils.summarise_species_protein_data(fasta_desc_lines)
yaml_text = yaml.dump(summary, explicit_start=True, default_flow_style=False)
with sys.stdout as fh:
fh.write(yaml_text)
In the code above we make use of the yaml module to convert our data structure
to the YAML file format. The PyYAML package is not part of the Python’s
standard library, but it is easily installed using pip (for more detailed
instructions see Installing Python packages).
$ sudo pip install pyyaml
The script also makes use of sys.stdin and sys.stdout to read from
the standard input stream and write to the standard output
stream, respectively. This means that we can pipe in the content to
our script and pipe output from our script.
For example to examine the YAML
output using the less pager one could use the command below.
$ gunzip -c data/uniprot_sprot.2015-11-26.fasta.gz | python fasta2yaml_summary.py | less
This immediately reveals that there are organisms in the SwissProt FASTA file that have few protein associated with them.
Note
Remember the benefits of using pipes was described in Creating a work flow using pipes.
---
AKT8 murine:
AKT8 murine leukemia virus: 1
AKV murine:
AKV murine leukemia virus: 3
Abelson murine:
Abelson murine leukemia virus: 3
Abies alba:
Abies alba: 1
Abies balsamea:
Abies balsamea: 3
Note
The formatting in the YAML file above was created for us by the call
to the yaml.dump() method in the fasta2yaml_summary.py
script.
Great work!
Key concepts
- Think about the problem at hand before you start coding
- Break down large tasks into smaller more manageable tasks, repeat until the tasks seem trivial
- Test-driven development is a software development practise that can help break tasks into manageable chunks that can be tested
- Many aspects of biological data processing boil down to string manipulations
- Regular expressions are a powerful tool for performing string manipulations, but use with caution as they can result in confusion
- Python has many built-in packages for performing complex tasks, in this chapter
we used the
repackage for working with regular expressions - There are also many third party Python packages that can be installed, in
this chapter we made use of the
yamlpackage for writing out a data structure as a YAML file - If in doubt, turn to the abundance of resources for Python online including manuals, tutorials and help forums